4. Tutorials¶
4.1. Demo: ChIP-seq analysis¶
4.1.1. Case: ChIP-seq study of Tbf1 in S. cerevisiae¶
Reference
Preti M, Ribeyre C, Pascali C, Bosio MC et al. The telomere-binding protein Tbf1 demarcates snoRNA gene promoters in Saccharomyces cerevisiae. Mol Cell 2010 May 28;38(4):614-20. PMID: 20513435
Abstract
Small nucleolar RNAs (snoRNAs) play a key role in ribosomal RNA biogenesis, yet factors controlling their expression are unknown. We found that the majority of Saccharomyces snoRNA promoters display an aRCCCTaa sequence motif at the upstream border of a TATA-containing nucleosome-free region. Genome-wide ChIP-seq analysis showed that these motifs are bound by Tbf1, a telomere-binding protein known to recognize mammalian-like T(2)AG(3) repeats at subtelomeric regions. Tbf1 has over 100 additional promoter targets, including several other genes involved in ribosome biogenesis and the TBF1 gene itself. Tbf1 is required for full snoRNA expression, yet it does not influence nucleosome positioning at snoRNA promoters. In contrast, Tbf1 contributes to nucleosome exclusion at non-snoRNA promoters, where it selectively colocalizes with the Tbf1-interacting zinc-finger proteins Vid22 and Ygr071c. Our data show that, besides the ribosomal protein gene regulator Rap1, a second telomere-binding protein also functions as a transcriptional regulator linked to yeast ribosome biogenesis.
Access link
- GEO series: GSE20870
4.1.1.1. Setup analysis environment¶
Here, we create a directory that will contain the raw data, the SnakeChunks library, the reference genome data and the results of the workflow(s) used.
We are going to use a global variable: $ANALYSIS_DIR.
ANALYSIS_DIR=$HOME/ChIP-seq_GSE20870
mkdir -p ${ANALYSIS_DIR}
cd ${ANALYSIS_DIR}
4.1.1.2. Download SnakeChunks¶
We are going to download the SnakeChunks library in the analysis directory. Another possibility would be to download SnakeChunks in a fixed place, and create a symlink to the analysis directory.
wget --no-clobber https://github.com/SnakeChunks/SnakeChunks/archive/4.1.2.tar.gz
tar xvzf 4.1.2.tar.gz
ln -s SnakeChunks-4.1.2 SnakeChunks
4.1.1.3. Download reference genome & annotations¶
Here, we are going to download the genome sequence and annotation files in the analysis directory. It is also possible to define a fixed location to store genomes and then create a symlink to it.
It can be useful to store all the genomes in one place, in order to avoid duplication of big files. Also, most mapping algorithms need to index the genome before proceeding with the alignment. This index needs only be done once, but it takes time and storage space, so it’s better to avoid duplicating it.
mkdir ${ANALYSIS_DIR}/genome
wget -nc ftp://ftp.ensemblgenomes.org/pub/fungi/release-30/fasta/saccharomyces_cerevisiae/dna/Saccharomyces_cerevisiae.R64-1-1.30.dna.genome.fa.gz -P ${ANALYSIS_DIR}/genome
wget -nc ftp://ftp.ensemblgenomes.org/pub/fungi/release-30/gff3/saccharomyces_cerevisiae/Saccharomyces_cerevisiae.R64-1-1.30.gff3.gz -P ${ANALYSIS_DIR}/genome
wget -nc ftp://ftp.ensemblgenomes.org/pub/fungi/release-30/gtf/saccharomyces_cerevisiae/Saccharomyces_cerevisiae.R64-1-1.30.gtf.gz -P ${ANALYSIS_DIR}/genome
gunzip ${ANALYSIS_DIR}/genome/*.gz
4.1.1.4. Download raw data¶
mkdir -p ${ANALYSIS_DIR}/data/GSM521934 ${ANALYSIS_DIR}/data/GSM521935
wget --no-clobber ftp://ftp-trace.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByRun/sra/SRR/SRR051/SRR051929/SRR051929.sra -P ${ANALYSIS_DIR}/data/GSM521934
wget --no-clobber ftp://ftp-trace.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByRun/sra/SRR/SRR051/SRR051930/SRR051930.sra -P ${ANALYSIS_DIR}/data/GSM521935
4.1.2. Workflow ‘import_from_sra’¶
The purpose of this workflow is to convert .sra files to .fastq files. The .sra format (Short Read Archive) is used by the GEO database, but for downstream analyses we need to dispose of fastq-formatted files. You can check out the glossary to find out more about file formats.
4.1.2.1. Workflow execution¶
If you have followed the previous steps, you have a file organization that looks like this:
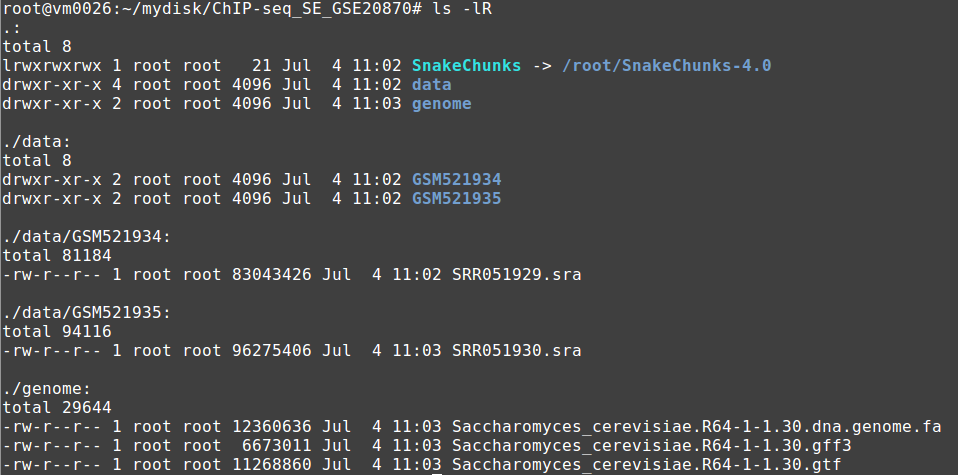
You should then be able to run the following command:
cd ${ANALYSIS_DIR}
snakemake -s SnakeChunks/scripts/snakefiles/workflows/import_from_sra.wf -p --configfile SnakeChunks/examples/ChIP-seq_SE_GSE20870/config.yml --use-conda
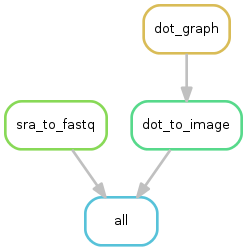
4.1.3. Workflow ‘quality_control’¶
This workflow can be run after the workflow ‘import_from_sra’, or directly on properly-organized fastq files (see this section if you dispose of your own data).
The purpose of this workflow is to perform quality check with FastQC.
Optionally, trimming can be performed using the tools Sickle or Cutadapt.
4.1.3.1. Workflow execution¶
cd ${ANALYSIS_DIR}
snakemake -s SnakeChunks/scripts/snakefiles/workflows/quality_control.wf -p --configfile SnakeChunks/examples/ChIP-seq_SE_GSE20870/config.yml --use-conda
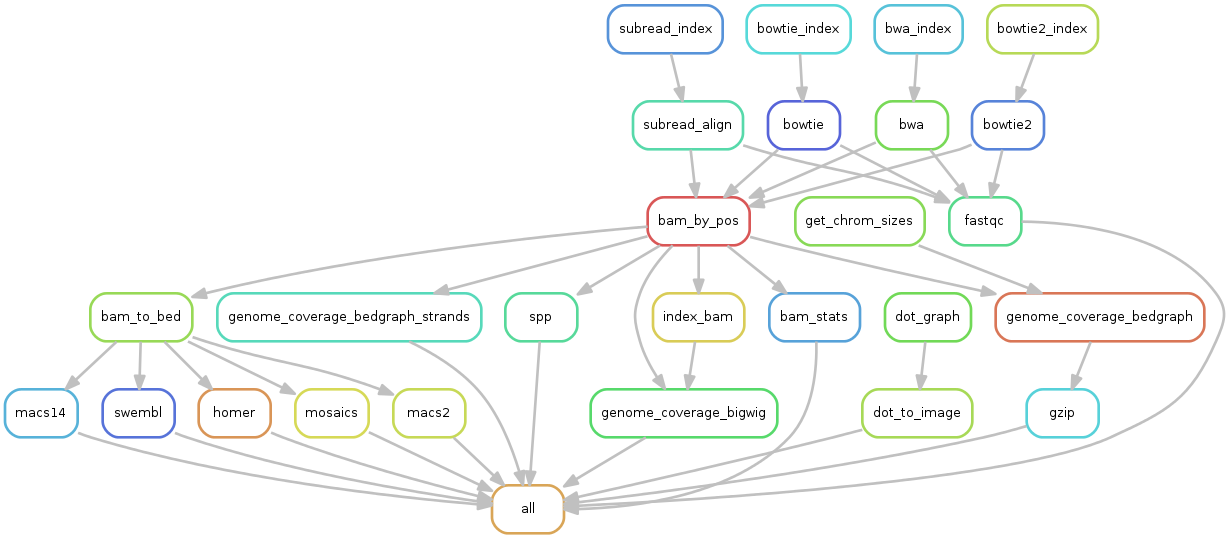
4.1.4. Workflow ‘ChIP-seq’¶
- This workflows performs:
- mapping with various algorithms
- genome coverage in different formats (check out our glossary)
- peak-calling with various algorithms
- motifs search using the RSAT suite
You must have run at least the workflow “import_from_sra”, and optionally the workflow “quality_control”.
4.1.4.1. Workflow execution¶
cd ${ANALYSIS_DIR}
snakemake -s SnakeChunks/scripts/snakefiles/workflows/ChIP-seq_peak-calling.wf -p --configfile SnakeChunks/examples/ChIP-seq_SE_GSE20870/config.yml --use-conda
4.2. Demo: ChIP-seq and RNA-seq integration¶
4.2.1. Case: Genomic analysis of the scc2-4 mutant in budding yeast¶
Reference
Genomic analysis of the scc2-4 mutant in budding yeast
Musinu Zakari
GEO series
4.2.2. Workflow ‘ChIP-seq’¶
ANALYSIS_DIR=$HOME/ChIP-seq_GSE55357
mkdir ${ANALYSIS_DIR}
cd ${ANALYSIS_DIR}
4.2.2.1. Download the SnakeChunks library¶
wget --no-clobber https://github.com/SnakeChunks/SnakeChunks/archive/4.1.2.tar.gz
tar xvzf 4.1.2.tar.gz
ln -s SnakeChunks-4.1.2 SnakeChunks
4.2.2.2. Download reference genome & annotations¶
wget -nc ftp://ftp.ensemblgenomes.org/pub/fungi/release-30/fasta/saccharomyces_cerevisiae/dna/Saccharomyces_cerevisiae.R64-1-1.30.dna.genome.fa.gz -P ${ANALYSIS_DIR}/genome
wget -nc ftp://ftp.ensemblgenomes.org/pub/fungi/release-30/gff3/saccharomyces_cerevisiae/Saccharomyces_cerevisiae.R64-1-1.30.gff3.gz -P ${ANALYSIS_DIR}/genome
wget -nc ftp://ftp.ensemblgenomes.org/pub/fungi/release-30/gtf/saccharomyces_cerevisiae/Saccharomyces_cerevisiae.R64-1-1.30.gtf.gz -P ${ANALYSIS_DIR}/genome
gunzip ${ANALYSIS_DIR}/genome/*.gz
4.2.2.3. Download ChIP-seq data¶
wget --no-clobber ftp://ftp-trace.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByRun/sra/SRR/SRR117/SRR1176905/SRR1176905.sra -P ${ANALYSIS_DIR}/data/GSM1334674
wget --no-clobber ftp://ftp-trace.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByRun/sra/SRR/SRR117/SRR1176907/SRR1176907.sra -P ${ANALYSIS_DIR}/data/GSM1334676
wget --no-clobber ftp://ftp-trace.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByRun/sra/SRR/SRR117/SRR1176908/SRR1176908.sra -P ${ANALYSIS_DIR}/data/GSM1334679
wget --no-clobber ftp://ftp-trace.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByRun/sra/SRR/SRR117/SRR1176910/SRR1176910.sra -P ${ANALYSIS_DIR}/data/GSM1334677
4.2.2.4. Workflow execution¶
Your directory should now look like this:
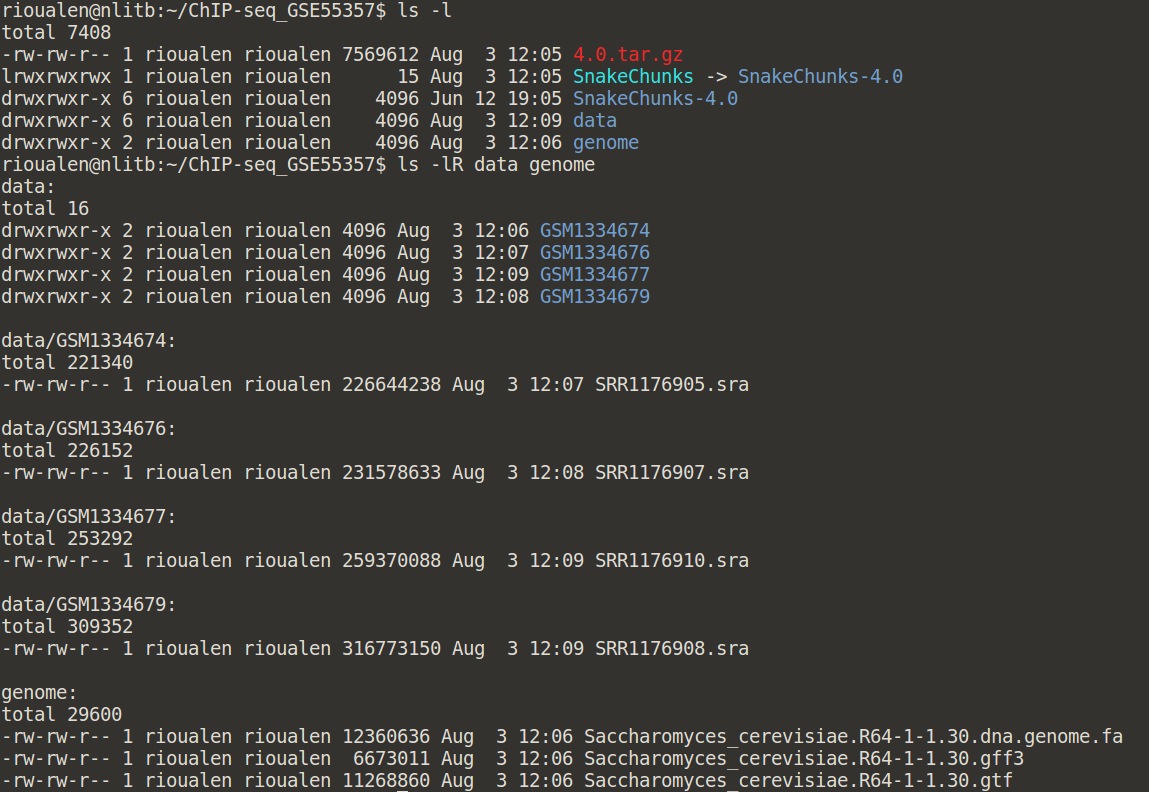
And you should be able to execute it like this:
cd ${ANALYSIS_DIR}
snakemake -s SnakeChunks/scripts/snakefiles/workflows/import_from_sra.wf -p --configfile SnakeChunks/examples/ChIP-seq_GSE55357/config.yml
snakemake -s SnakeChunks/scripts/snakefiles/workflows/quality_control.wf -p --configfile SnakeChunks/examples/ChIP-seq_GSE55357/config.yml
snakemake -s SnakeChunks/scripts/snakefiles/workflows/ChIP-seq_peak-calling.wf -p --configfile SnakeChunks/examples/ChIP-seq_GSE55357/config.yml
4.2.3. Workflow ‘RNA-seq’ for differential expression analysis¶
ANALYSIS_DIR=$HOME/RNA-seq_GSE55316
mkdir ${ANALYSIS_DIR}
cd ${ANALYSIS_DIR}
4.2.3.1. Download the SnakeChunks library¶
wget --no-clobber https://github.com/SnakeChunks/SnakeChunks/archive/4.1.2.tar.gz
tar xvzf 4.1.2.tar.gz
ln -s SnakeChunks-4.1.2 SnakeChunks
4.2.3.2. Download reference genome & annotations¶
wget -nc ftp://ftp.ensemblgenomes.org/pub/fungi/release-30/fasta/saccharomyces_cerevisiae/dna/Saccharomyces_cerevisiae.R64-1-1.30.dna.genome.fa.gz -P ${ANALYSIS_DIR}/genome
wget -nc ftp://ftp.ensemblgenomes.org/pub/fungi/release-30/gff3/saccharomyces_cerevisiae/Saccharomyces_cerevisiae.R64-1-1.30.gff3.gz -P ${ANALYSIS_DIR}/genome
wget -nc ftp://ftp.ensemblgenomes.org/pub/fungi/release-30/gtf/saccharomyces_cerevisiae/Saccharomyces_cerevisiae.R64-1-1.30.gtf.gz -P ${ANALYSIS_DIR}/genome
gunzip ${ANALYSIS_DIR}/genome/*.gz
4.2.3.3. Download RNA-seq data¶
wget --no-clobber ftp://ftp-trace.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByRun/sra/SRR/SRR117/SRR1176894/SRR1176894.sra -P ${ANALYSIS_DIR}/data/GSM1334027
wget --no-clobber ftp://ftp-trace.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByRun/sra/SRR/SRR117/SRR1176896/SRR1176896.sra -P ${ANALYSIS_DIR}/data/GSM1334029
wget --no-clobber ftp://ftp-trace.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByRun/sra/SRR/SRR117/SRR1176900/SRR1176900.sra -P ${ANALYSIS_DIR}/data/GSM1334033
wget --no-clobber ftp://ftp-trace.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByRun/sra/SRR/SRR117/SRR1176901/SRR1176901.sra -P ${ANALYSIS_DIR}/data/GSM1334034
4.2.3.4. Workflow execution¶
Your directory should now look like this:
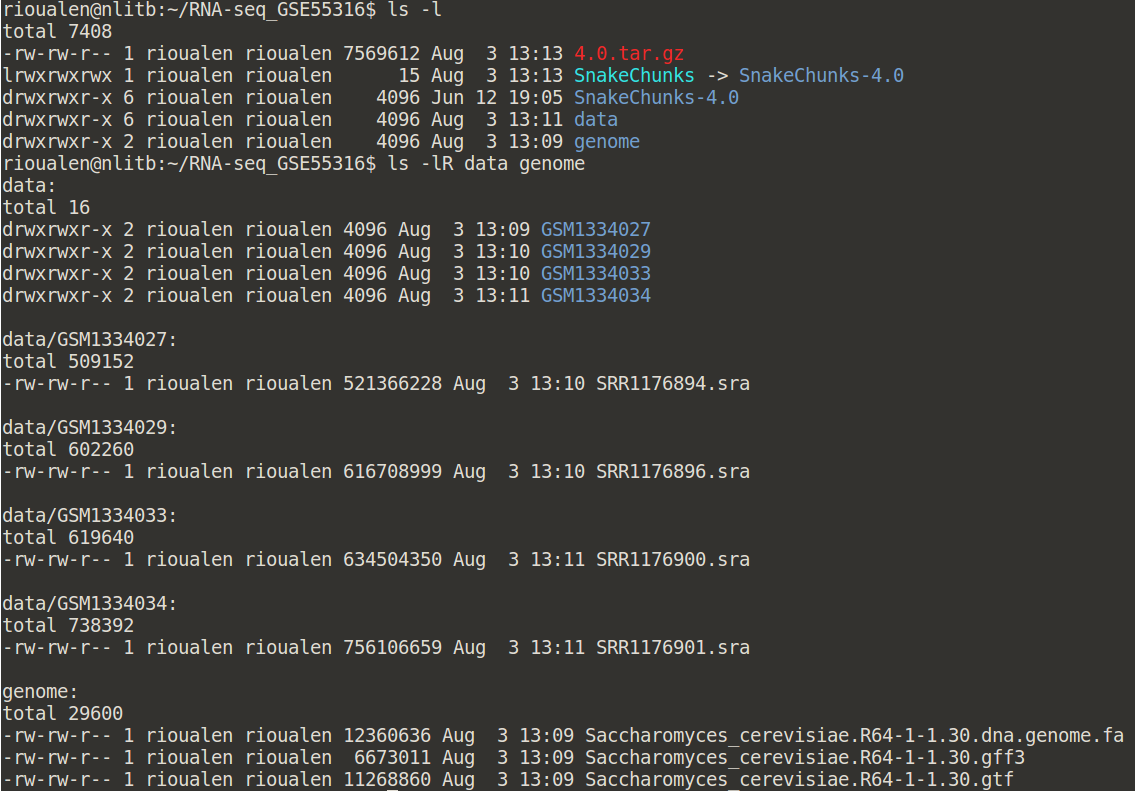
And you should be able to execute it like this:
cd ${ANALYSIS_DIR}
snakemake -s SnakeChunks/scripts/snakefiles/workflows/import_from_sra.wf -p --configfile SnakeChunks/examples/RNA-seq_GSE55316/config.yml
snakemake -s SnakeChunks/scripts/snakefiles/workflows/RNA-seq_complete.wf -p --configfile SnakeChunks/examples/RNA-seq_GSE55316/config.yml
4.2.4. Workflow ‘integration_ChIP_RNA’¶
coming soon
4.3. Demo: alternative transcripts with RNA-seq¶
4.3.1. Case: C.elegans¶
4.3.1.1. Setup workdir¶
ANALYSIS_DIR=$HOME/GSE59705_RNA-seq_splicing
mkdir ${ANALYSIS_DIR}
cd ${ANALYSIS_DIR}
4.3.1.2. Download the SnakeChunks library¶
wget --no-clobber https://github.com/SnakeChunks/SnakeChunks/archive/4.1.2.tar.gz
tar xvzf 4.1.2.tar.gz
ln -s SnakeChunks-4.1.2 SnakeChunks
4.3.1.3. Download reference genome & annotations¶
wget -nc ftp://ftp.wormbase.org/pub/wormbase/releases/WS220/species/c_elegans/c_elegans.WS220.genomic.fa.gz -P ${ANALYSIS_DIR}/genome
wget -nc ftp://ftp.wormbase.org/pub/wormbase/releases/WS220/species/c_elegans/c_elegans.WS220.annotations.gff3.gz -P ${ANALYSIS_DIR}/genome
wget -nc ftp://ftp.wormbase.org/pub/wormbase/releases/WS253/species/c_elegans/PRJNA13758/c_elegans.PRJNA13758.WS253.canonical_geneset.gtf.gz -P ${ANALYSIS_DIR}/genome
gunzip ${ANALYSIS_DIR}/genome/*.gz
wget -nc ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR152/001/SRR1523361/SRR1523361_2.fastq.gz -P ${ANALYSIS_DIR}/fastq/GSM1443914
wget -nc ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR152/001/SRR1523361/SRR1523361_1.fastq.gz -P ${ANALYSIS_DIR}/fastq/GSM1443914
wget -nc ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR152/002/SRR1523362/SRR1523362_2.fastq.gz -P ${ANALYSIS_DIR}/fastq/GSM1443915
wget -nc ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR152/002/SRR1523362/SRR1523362_1.fastq.gz -P ${ANALYSIS_DIR}/fastq/GSM1443915
wget -nc ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR152/003/SRR1523363/SRR1523363_2.fastq.gz -P ${ANALYSIS_DIR}/fastq/GSM1443916
wget -nc ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR152/003/SRR1523363/SRR1523363_1.fastq.gz -P ${ANALYSIS_DIR}/fastq/GSM1443916
wget -nc ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR152/004/SRR1523364/SRR1523364_2.fastq.gz -P ${ANALYSIS_DIR}/fastq/GSM1443917
wget -nc ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR152/004/SRR1523364/SRR1523364_1.fastq.gz -P ${ANALYSIS_DIR}/fastq/GSM1443917
gunzip ${ANALYSIS_DIR}/fastq/*/*.gz
- rename
snakemake -s SnakeChunks/scripts/snakefiles/workflows/RNA-seq_transcripts.wf -p --configfile SnakeChunks/examples/RNA-seq_GSE59705/config.yml -n
4.4. Running SnakeChunks workflows on your own data¶
4.4.1. SnakeChunks library & genome data¶
Hereafter is a suggestion for the organization of your files.
ANALYSIS_DIR=$HOME/my_analysis
mkdir -p ${ANALYSIS_DIR}
cd ${ANALYSIS_DIR}
# Download the SnakeChunks library
wget --no-clobber https://github.com/SnakeChunks/SnakeChunks/archive/4.1.2.tar.gz
tar xvzf 4.1.2.tar.gz
ln -s SnakeChunks-4.1.2 SnakeChunks
# Download genome data
wget -nc <URL_to_my_genome.fa.gz> -P ${ANALYSIS_DIR}/genome
wget -nc <URL_to_my_genome.gff3.gz> -P ${ANALYSIS_DIR}/genome
wget -nc <URL_to_my_genome.gtf.gz> -P ${ANALYSIS_DIR}/genome
gunzip ${ANALYSIS_DIR}/genome/*.gz
Your directory should look like this:
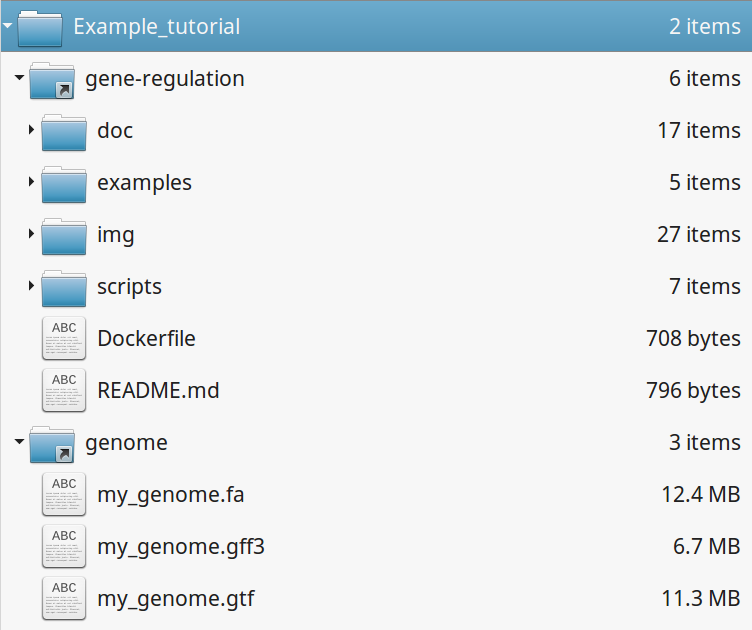
4.4.2. Fastq files organization¶
This tutorial assumes you dispose of your own fastq files. We recommend that your organize your samples in separate folders, and name both fastq files and their parent directories accordingly.
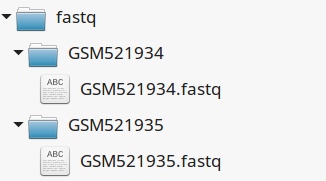
If you have paired-ends samples, they should be in the same directory and distinguished using a suffix of any sort.
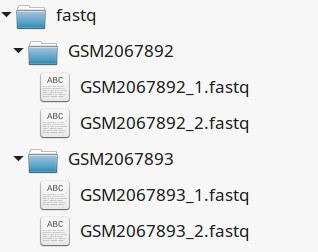
4.4.3. Metadata¶
Running the workflows provided by the SnakeChunks library requires the use of three configuration files.
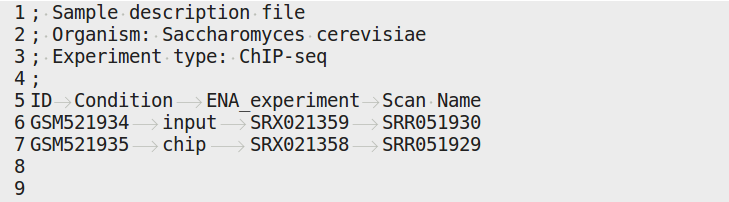
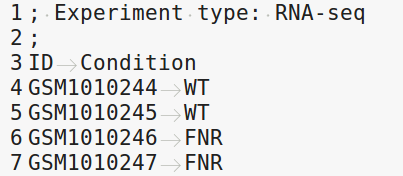
4.4.3.1. samples.tab¶
This file should contain, at least, one column named “ID”, that should contain sample names matching those defined in the previous section. In the case of an RNA-seq analysis, it should also contain a column “Condition”, which will define groups of comparison (see design file in the section below).
All the samples will be processed in the same manner. You can prevent certain samples from being processed by commenting the corresponding lines with a “;” at the beginning of the line.
RNA-seq sample groups should contain at least 2 samples.
You can add any other relevant information related to samples in other tab-separated columns.
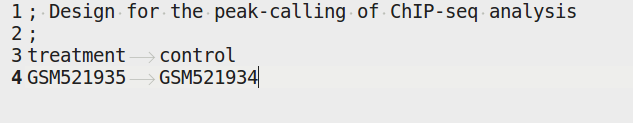
4.4.3.2. design.tab¶
The purpose of this file is to determine which samples should be processed together. In a ChIP-seq analysis, it will be used to define which ChIP samples should be compared with which inputs. In an RNA-seq experiment, it defines the conditions to be compared against each other.
Column names should be respected.

4.4.3.3. config.yml¶
You can find examples of configuration files in the examples section of the SnakeChunks directory.
Directories should be defined relative to the working directory defined in the beginning: genome, SnakeChunks, fastq, etc. Same goes for configuration files.
Genome filenames should be mentionned as they appear in the defined genome directory.
Genome size should be filled in, as well as the sequencing type: “se” for single-end data, and “pe” for paired-ends data. In the case of paired-ends data, suffixes (parameter “strands”) should be mentioned and should match the filenames (minus the “_”).
The minimum of configuration should look like this:

All the parameters related to the tools used are optional, and the default parameters of each program will be used when they’re not set in the configfile.
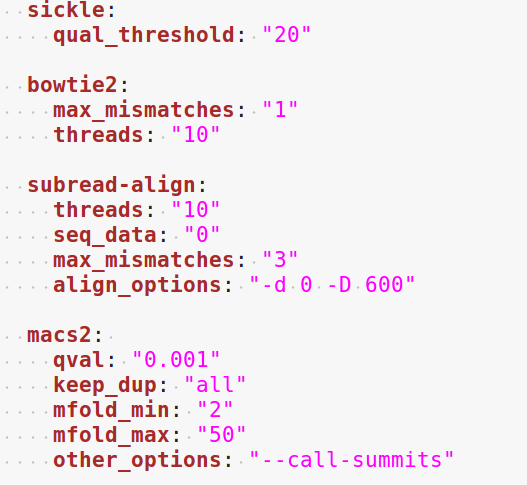
4.4.4. Running a workflow¶
If your directory now looks like this, you should be ready to run a worflow!
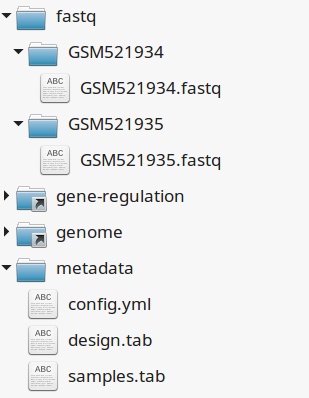
You can verify it by doing dry runs:
cd ${ANALYSIS_DIR}
# Run the quality check
snakemake -s SnakeChunks/scripts/snakefiles/workflows/quality_control.wf --config-file metadata/config.yml -p -n
# Run the ChIP-seq workflow
snakemake -s SnakeChunks/scripts/snakefiles/workflows/ChIP-seq.wf --config-file metadata/config.yml -p -n
# Run the RNA-seq workflow
snakemake -s SnakeChunks/scripts/snakefiles/workflows/RNA-seq.wf --config-file metadata/config.yml -p -n
Just remove the -n option to actually run them.